
Research Article | DOI: https://doi.org/10.31579/2768-0487/012
1Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Libya.
2 Judicial Expertise and Research Centre, Tripoli, Libya.
3Zoology Department, Faculty of Science, Tripoli University, Libya.
4Department of Biosciences, University of Salzburg, Salzburg, Austria
*Corresponding Author: Abdul M Gbaj, Professor of Genetics and Biochemistry, Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Libya.
Citation: I A Sadawe, N H Meiqal, S M Bensaber, A R Hamid. Abdul M G. (2021) Estimation of CYP3A4*1B single nucleotide polymorphism using target-assembled in-situ detection by synthetic DNA-mounted excimers. Journal of Clinical and Laboratory Research. 2(2) DOI: 10.31579/2768-0487/012
Copyright: ©2021 Abdul M Gbaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 27 February 2021 | Accepted: 26 March 2021 | Published: 30 March 2021
Keywords: dna probe systems; excimers; dna detection; fluorescence; cyp3a4*1b; stokes shift
Abstract
CYP3A4*1B is a single nucleotide polymorphism of CYP3A4 and is associated with prostate cancer which exhibits higher nifedipine oxidase activity in liver. This research provides details of the effects of structural variation and medium effects for the recently reported split-oligonucleotide (tandem) probe system for excimers-based fluorescence detection of DNA. In this approach the detection system is split at a molecular level into signal-silent components, which must be assembled correctly into a specific 3-dimensional structure to ensure close proximity of the excimer partners and the consequent excimer fluorescence emission on excitation. The model system consists of two 11-mer oligonucleotides, complementary to adjacent sites of a 22-mer DNA target. Each oligonucleotide probeis equipped with functions able to form an excimer on correct, contiguous hybridization. The extremely rigorous structural demands for excimer formation and emission required careful structural design of partners for excimer formation, which are here described. This study demonstrates that the excimer formed emitted at ~480 nm with alarge Stokes shift (~130 - 140 nm).
Reversible hybridisation of complementary polynucleotides is esasential to the biological processes of replication, transcription, and translation. Physical studies of nucleic acid hybridisation are required for understanding these biological processes on a molecular level. The physical characterisation of nucleic acid hybridisation is essential for predicting the performance of nucleic acids in vitro, for instance, in hybridisation assays used to detect specific polynucleotide sequences.
Fluorescence measurements present an improved sensitive measure of nucleic acid concentration compared to conventional solution-phase detection techniques. Additionally, the sensitivity of fluorophores to their environments offers a means by which to differentiate hybridised from unhybridised nucleic acids without resorting to separation techniques. This was first demonstrated by attaching different fluorescent labels to the termini of oligonucleotides, which hybridise to adjacent regions on a complementary strand of DNA. Appropriate selection of fluorophores led to adetectable signal between the labels on hybridisation of the two-labeled strands to their complementary strand. For example, split-probe systems based on excimer fluorescence were first described by Ebata et al.[1-3], who attached pyrene to the 5'-terminus of one oligonucleotide probe and to the 3'-terminus of the other oligonucleotide probe. The probes bound to adjacent regions of the target, bringing the pyrene molecules into close proximity, forming an excimer[4, 5]. Excimer emission from oligonucleotides containing 5-(1-pyrenylethynyl)uracil [6], trans-stilbene [7], and perylene [8] have also been reported.
Numerous genetic diseases have been found to result from a change of a single DNA base pair. These single nucleotide polymorphisms (SNP) may cause changes in the amino acid sequence of important proteins[9, 10]. Methods sensitive to single base-pair mutations for the fast screening of patient samples to identify disease-causing mutations will be essential for diagnosis, prevention and treatment. Usually hybridisation analysisis used, where a short, probe oligonucleotide (15-20 base pairs) bearing some kind of label (e.g. fluorophore) hybridises to complementary base pairs in DNA or RNA. The nucleic acids required for analysis can be recovered from a variety of biological samples including blood, saliva, urine, stool, nasopharyngeal secretions or tissues[11-13]. Highly specific, simple, and accessible methods are needed to meet the accurate requirements of single nucleotide detection in pharmacogenomic studies, linkage analysis, and the detection of pathogens. Recently there has been a move away from radioactive labels to fluorescence.
It has been reported[14] that an emissive exciplex can be formed by juxtaposition of two differentexternally oriented exciplex-forming partners (pyrene and naphthalene) at the interface (nick region) of tandem oligonucleotides forming a duplex of some kind on hybridization with their complementary target strand. We have been mainly interested in using excimer fluorescence signals to study the hybridisation between two fluorophore-labeled complementary DNA strands, as shown in Figure 1. Attaching fluorescent labels (pyrene and pyrene in Figure 1) to the probe of complementary DNA strands showed strong interactions between particular fluorophore pairs on hybridisation.Split-probe systems based on two 11mer probe strands were investigated in this paper using the base sequences shown in Figure 1.
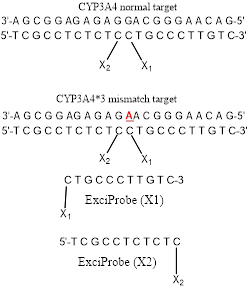
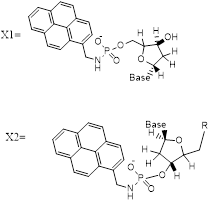
In the split-probe model system (Figure 1) two 11-mer probe oligonucleotides labelled with 1-pyrenylmethylamine (Pyrene) attached to 3′ and 5′ terminal phosphate groups. Hybridization of these probes to a complementary 22-mer oligonucleotide target resulted in correct orientation of the two pyrenes excimer-partners. Excitation of the pyrenylmethylamino partner at 350 nm led to the structure of an excited-state complex (excimer) with the pyrene partner. This excimer emitted at a longer wavelength of 480 nm (Stokes shift 130 nm) as compared with a mixture of unhybridisedsplit-probes. The excimer emission was particularlypreferred by the use of trifluoroethanol as co-solvent (80 % v/v)[14].
The split-probe model system used in this study containing 22-mer target sequence which corresponds to a region of the CYP3A4 major genome (3'-AGCGGAGAGAGGACGGGAACAG-5') and complementary 11mer probes(5'-TCGCCTCTCTC-pyrene and pyrene-CTGCCCTTGTC-3'). CYP3A4*1B (-392A>G, rs2740574) is a CYP3A4 polymorphism and it is the frequently studied proximal promoter variant which occurs in White humanpopulations at around 2–9% but at elevated frequencies in Africans including Libyans[15-17]. We now report how this excimerstrategy can permit detection of an allelic variant of the human CYP3A4*1B gene sequence.
Single-nucleotide polymorphisms (SNPs) in genes coding for cytochrome P450 (CYP) enzymes have been linked to many diseases and to inter-individual differences in the efficiency and toxicity of many drugs. Thirty seven CYP3A4 variants, with amino-acid changes located in coding regions, have been identified among the different ethnic populations (www.pharmvar.org/gene/CYP3A4). For example, CYP3A4*1B allele (CYP3A4-V, rs2740574), a −392A>G transition in the promoter region, has been reported to be considerably connected with HIV infection[18], increased threat of hormone negative breast cancer(missing estrogen, progesterone receptors)[19], prostate cancer [20]and increased risk for developing leukemia after epipodophyllotoxin therapy[21]. Also, theCYP3A4*1B allele causes amino acid substitution affectingthe metabolism of a range of drugs such as Nifidipine and Carbamazepine which leads to altered enzyme activity and drug sensitivity, e.g. the mutant enzyme results in impaired metabolism[22, 23].
The excimer constructs used standard DNA base / sugar structures in both complementary probes. The targetswere a part of the CYP3A4 chromosome 7 sequence band q22.1 (Genbankcode[ENSG00000160868 nucleotides r=7:99354604-99381888]: 3'-AGCGGAGAGAGGACGGGAACAG-5' (the bold base provided the SNP location G>A). The ExciProbes had the sequence (X1) p-5'-CTGCCCTTGTC-3' and (X2) 5'-TCGCCTCTCTC-3'-p. The probes were supplied with a free 3’ or 5’-phosphate group (p). Reagents of the highest quality available and DNA probes and DNA targetswere purchased from Sigma-Aldrich(Paris France,). Distilled water wasfurtherpurified by ion exchange and charcoal using a MilliQ system (Millipore Ltd, UK). Tris buffer was prepared from analytical reagent grade materials. pH was measured using a Hanna(Lisbon, Portugal)HI 9321 microprocessor pH meter, calibrated with standard buffers (Sigma-Aldrich) at 20 °C.
HPLC
HPLC purification of probes was performed on an Agilent 1100 Series HPLC system (California, USA), consisting of a quaternary pump with solvent degasser, a diode-array module for multi-wavelength signal detection using an Agilent 1100 Series UV-visible detector and an Agilent 1100 Series fluorescence detector for on-line acquisition of excitation/emission spectra. The system had a manual injector and thermostatted column compartment with two heat exchangers for solvent pre-heating. The HPLC system was operated by Agilent HPLC 2D ChemStation Software. Depending on the purification performed, the columns used were: Zorbax Eclipse X DB-C8 column(California, USA) (length 25 cm, inner diameter 4.6 mm, particle size 5μm), or a Luna C18 (2) column (California, USA) (length 25 cm, inner diameter 4.6 mm, particle size 5 μm) with elution using an increasing gradient (0–50%) of acetonitrile in water (fraction detection at 260, 280, and 340 nm).
UV-visible spectrophotometry
UV–visible absorption spectra were measured at 20°C on a Cary-Varian 1E UV–visible spectrophotometer (London, UK.) with a Peltier-thermostatted cuvette holder and Cary 1E operating system/2 (version 3) and CARY1 software. Quantification of the oligonucleotide components used millimolar extinction coefficients (e260) of 99.0 for ExciProbe (X1), 94.6 for ExciProbe (X2). The extinction coefficients were calculated by the nearest neighbour method [24] and the contribution of the exci-partners was neglected.
Spectrophotofluorimetry
Fluorescence emission/excitation spectra were recorded in 4-sided quartz thermostatted cuvettes using a Peltier-controlled-temperature Cary-Eclipse, spectrofluorophotometer (London, UK). All experiments were carried out at 5°C. Hybridisation: Duplex formation was induced by sequential addition of ExciProbe(X1)andExciProbe (X2). The mole ratio of all oligonucleotides ExciProbe (X1)and ExciProbe (X2) used were 1:1, the concentration of each component was2.5 µM. Tris buffer was added either with or without 80% TFE and thevolume made up to 1000 µl with deionisedwater. Excitation wavelengths of 340 nm (for the pyrene monomer) and 350 nm (for the full two probes and the target) were used, at slit width of5 nm and recorded in the range of 350-650 nm. Emission spectra were recorded after each sequential addition of each component to record the change in emission of each addition. A baseline spectrum of buffer and water or buffer, water and 80% TFE was always carried out before start of the measurement. After each addition the solution was left to equilibrate for 6 minutes in the fluorescence spectrophotometer and emission spectra wererecorded until no change in the fluorescence spectra was seen to ensure it had been reached.The sequence of experiments was first using ExciProbe (X1) then ExciProbe (X2). Control experiments were conducted using firstly ExciProbe (X1) followed bythe 3’-free oligonucleotide probe and finally the complementary target. Allspectra were buffer corrected.
Control experiments
Control experiments were carried out in 80% TFE/Tris buffer as for the experimental systems using the standard method described above. The control experimentwas performed toconfirm whether the obtained excimer emission is a result of such background effects or arise from the hypothesised excimer structures. Then fluorescence melting curve experiments (based on excitation 350 nm and emission 480 nm for the excimer) were performed using a Cary Eclipse fluorescence spectrophotometer by measuring the change in fluorescence intensity for the excimer with meltingtemperature(Tm). Tm was also determined spectrophotometrically by measuring the change in absorbance at 260 nm with temperature. Tm was determined either by taking the point at half the curve height or using the first derivative method.
Synthesis and oligonucleotide modification
Attachments of 1-pyrenemethylamine to oligonucleotide probes were as describedin[14, 25]. One equivalent of 1-pyrenemethylamine was attached via phosphoramide links to the terminal 5’-phosphate of (X1) p-5'-CTGCCCTTGTC-3' probe and to the 3’-phosphate of (X2) 5'-TCGCCTCTCTC-3'-p. To the cetyltrimethylammonium salts of the oligonucleotides (~1micromole) dissolved in N, N-dimethylformamide (200 l) were added triphenylphosphine (80 mg, 300 mol) and 2,2'-dipyridyl disulfide (70 mg, 318 mol), and the reaction mixture was incubated at 37 C for 10 min. 4-N’,N’-Dimethylaminopyridine (40 mg, 329 mol) was added, the reaction mixture incubated for a further 12 minutes at 37 ºC and 1-pyrenemethylamine hydrochloride (4 mg, 14.9 μmol, dissolved in 100 l of N, N-dimethylformamide and three microliter triethylamine) added. The mixture of the reaction was incubated at 37 ºC for full day (24 hours). product then was purified using reverse-phase HPLC (eluted by 0.05 M LiClO4 with a gradient from 0 to 60 % acetonitrile).
CYP3A4*1B single nucleotide polymorphism
Split-probe systems were used to investigate the effect of SNP in the CYP3A4*1B target sequence on excimer emission compared to the normal-type target. Experiments were carried out in 80% TFE/Tris buffer at 5°C. The sequence of addition was: ExciProbe (X1), ExciProbe (X2), and finally 22 mer mutant-target oligonucleotide (CYP3A4*1B). All spectra were buffer-corrected.
Excimer formation using terminally located probe systems
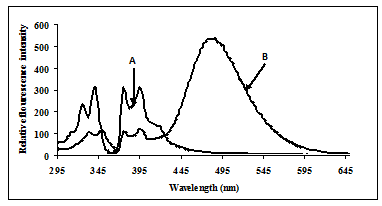
Fluorescence studies were made for solutions of ExciProbe (X1) and ExciProbe (X2) oligonucleotides with both probes complementary to each other (Figure 1). Figure 2 shows the excitation and emission spectra for (A) the ExciProbe (X1) and ExciProbe (X2) in 80% TFE/Tris buffer (0.01 M Tris, 0.1 M NaCl, pH 8.4), at5ºC, (B) ExciProbe (X1) and ExciProbe (X2) hybridised to the 22-mer target oligonucleotide. On excitation at 350 nm, the 3'-pyrenyl ExciProbe (X1) and 5'-pyrenyl ExciProbe (X2) showed fluorescence typical of pyrene LES emission (lmax = 376, 395 nm). Addition of the complementary target resulted in immediate quenching of the LES emission at 395 nm to less than one-third of its original value and the appearance of a new, broad emission band (lmax = 480 nm) characteristic of pyrene excimer fluorescence after the full terminally located system had formed)[1, 2, 26]. Addition of the two probes to the target also caused a slight red shift in both excitation (from 342 nm to 349 nm) and emission (from 376 nm to 378 nm; λex 350 nm) spectra, consistent with duplex formation[1, 2, 26].
Control experiments for theterminally located excimer system
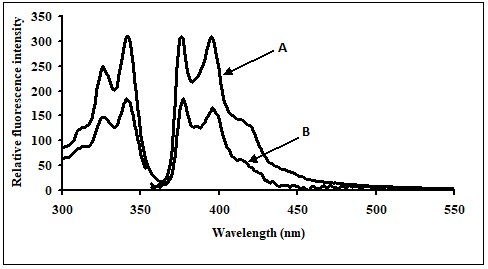
Control experiments on a 1:1 mixture of 5'-pyrenyl ExciProbe (X1) and 3'-pyrenyl ExciProbe probe (X2) oligonucleotides were carried out in 80% TFE/0.01 M Tris, 0.1 M NaCl, pH 8.4 to determine if the fluorescence was from pyrene interacting as an excimer with the intended pyrene exci-partner, or an interaction with bases of the oligonucleotides. The 5'-pyrenyl ExciProbe (X1) showed no band at 480 nm in the absence of the target oligonucleotide (Figure3). Addition of the complementary oligonucleotide target to ExciProbe (X1) resulted in a slight shift in λmax of LES emission to 379 nm, consistent with hybridisation of the probe with the complementary target. However, no marked 480 nm band was seen, even after heating the system to 70ºC and re-annealing by slowly cooling back to 5ºC. The weak fluorescence emission at 480 nm for the control duplex (before and after heating cooling, Figure 3) on duplex formation appeared real and could be related to exciplex formation, due to intra-molecular interaction of pyrene within the assembled duplex. However, relative to the full system with both 3'- and 5'-pyrenyl groups (Figure 3) the emission at 480 nm is insignificant.
CYP3A4*1B single nucleotide polymorphism
The excimer emission was detected (broadband at ~ 480 nm) for normal target and showed strong emission at 480 nm (545 relative fluorescence intensity) compared to the mutated target (220 relative fluorescence intensity) around 2.5 fold (Figures 4).

Melting temperatures of SNP
Melting curve experiments were performed spectrophotometrically at A260 and estimated by using the first derivative method. The melting temperatures (Tm) for normal CYP3A4 target was 76.9 ±0.8°C and 75.0 ±0.8°C for CYP3A4*1B, respectively. The melting temperature at 260 nm for systems was performed in 80% TFE/Tris buffer (10 mM Tris, 0.1 M NaCl, pH 8.4). Control experiments for Tm were carried out in 80% TFE/Tris buffer. In addition, similar thermal results were obtained using fluorescence melting curve experiments based on excitation 350 nm and emission 480 nm for the excimer. The fluorescence thermal study was performed using a Cary Eclipse fluorescence spectrophotometer by measuring the change in fluorescence intensity for the excimer with temperature.
Confirmation of duplex formation
In our experimentsofhybridising the two 11 mer probes to the complementary target in phosphate buffer (pH 7.0) containing 0.1 M NaCI, the pyrene moieties of the two probes came into close proximity, and an excimer band at 480 nm was generated. This result is consistent with results obtained by Yamanaet al.(1994) who used a system that incorporated a pyrene-modified nucleotide at the 5'-end of one probe and a pyrene-modified nucleotide at the 3'-end of the other[27]. Figure2 shows fluorescence typical of pyrene local excited state (LES) emission (lmax = 376, 395 nm) for a 5'-pyrenyl ExciProbe (X1) labelled oligonucleotide alone. The emission spectrum obtained is similar to that obtained in the literature using10 mM phosphate buffer (pH 7.0) 20 % v/v DMF, 0.2 M NaCl at 25°C and gave lmax = 377, 396 nm [1, 2, 26]. Addition of the 5'-pyrenyl ExciProbe (X1) to the 3'-pyrenyl ExciProbe (X2) target resulted in immediate quenching of the LES emission at 395 nm to less than one-third of its original value and the appearance of a new, broad emission band atlmax = 480 nm characteristic of pyrene excimer fluorescence (Figure 2).
Melting experiments provide further strong evidence of duplex formation. The split-probe systems showed sigmoid single-transition melting curves spectrophotometrically (A260 or A350) or spectrofluorometrically from fluorescence intensity at 340 nm for the LES (lex) and 376 nm (lem) for thepyrene monomer and at 350 nm for LES (lex) and 480 nm (lem) for the excimer (data not shown). Additional evidence of duplex formation comes from the emission spectra, as one probe oligonucleotide alone did not give an excimer signal in the absence of the other complementary probe.Further evidence of duplex formation and reversibility came from experiments using a heating and cooling cycle. Experiments of terminally located probe systems at different temperatures showed that the excimer intensity decreased when the temperature increased and eventually disappeared. This process is reversible, providing further evidence of duplex formation. A better-formed duplex structure probably enables the exci-partners to be better positioned for excimer formation. The reappearance of the excimer spectra on re-cooling indicates that no destruction of the components occurs on heating the system.
Evidence of excimer formation
The red-shifted structureless band at ~ 480 nm is characteristic of excimer emission, but could be due to interaction of the exci-partners with each other or nucleobases as pyrene are able to form an exciplex with certain nucleotide bases, especially guanine and to a lesser extent thymidine[28, 29]. Also some oligonucleotide sequences show weak exciplex emission from pyrene attached to their 5'-termini in the absence of any added (complementary) oligonucleotide[30]. Thus, it is important to establish for the terminally located system the origin of the emission at 480 nm.Heating the system caused the excimer emission intensity to decrease due to dissociation of the duplex structure. On re-cooling the system excimer emission reappeared. The Tm values by fluorescence and UV-visible methods were similar and of the magnitude expected for such a system (22-mer duplex)[31].
CYP3A4*1B single nucleotide polymorphism
The search for sequences that differ in only one or two nucleobases needs tools to detect nucleic acid sequences that have high performance, speed, simplicity, and low cost. There have been many different techniques developed to identify the mutations in nucleic acid sequences. Techniques based on matched/mismatched-duplex stabilities, restriction cleavage, ligation, nucleotide incorporation, mass spectrometry and direct sequencing have been reviewed[32, 33].The DNA split-probe system of CYP3A4*1B was able to discriminate between perfectly matched CYP3A4 and mismatched CYP3A4*1B targets. Several split-oligonucleotide systems have been reported to discriminate between SNPs. These include the ligation method of Landegenet al. [34], nanoparticle probes[35, 36] and the template-directed ligation method[37, 38]. The split-probe excimer system of Paris et al. [39] was found to be sensitive to a single-base mutation in the target, positioned four base pairs from the 3'-junction. In the Paris study the addition of the unmutated target to the pyrene probes resulted in an increase in 490 nm emission as well as a 4.7-fold decrease in 398 nm monomer emission. The resulting excimer:monomer ratio was 0.04, very different to that for the sequence with a single-base point mutation which was 2.7[39].
In the present study the duplexes containing GAGAACG/CTCCTGC mismatch is significantly destabilized compared with its correctly paired parent. Amber and Znosko[40]studied the thermodynamics of A/G mismatches in different nearest-neighbour contexts. They found a penalty (energy loos) of 1.2 kcal/mol for replacing a G-C base pair with either an A-U or G-U base pair.For both CYP3A4 (normal target) and CYP3A4*1B (mismatched target) showed a sigmoidal melting profile, typical of the dsDNA to ssDNA transition, providing further evidence of tandem duplex formation. The Tm values of CYP3A4*1B are less to those of the fully matched,consistent with literature studies performed on different sequences under identical conditions[25]. Duplexes of CYP3A4*1B (mismatched target) with mismatchesof G/A in the twelve position from the 3' and 5' ends, respectively, showed significantly lower Tm than CYP3A4 (normal target). These results indicate that the ∆G contribution of a single G/A mismatch and the position of the mismatch are crucial to duplex stabilityand consistent with the literature [41, 42]. The ∆G contribution of a single G/A mismatch to duplex stability was studied by Allawi et al. [43] who found that ∆G is dependent on the neighbouring base pairs and ranges from +1.16 kcal/mol (for the context TGA/AAT) to -0.78 kcal/mol (for the context GGC/CAG). Allawi et al. also showed that the nearest neighbour model is applicable to internal G/T mismatches in DNA. In their study of G/T mismatches, the most stable trimer sequence containing a G/T mismatch was -1.05 kcal/mol for CGC/GTG and the least stable was +1.05 kcal/mol for AGA/TTT. On average, when the closing Watson-Crick pair on the 5' side of the mismatch is an A/T or a G/C pair, G/A mismatches are more stable than G/T mismatches by about 0.40 and 0.30 kcal/mol, respectively [43, 44]. When the 5' closing pair is a T/A or a C/G, then G/T mismatches are more stable than G/A mismatches by 0.54 and 0.75 kcal/mol, respectively. Evidently, the different hydrogen-bonding and stacking in G/T and G/A mismatches results in different thermodynamic trends and the energy and structural information are the compositions of the following variables, such as bond angle energies, bond energies, planarity energies, dihedral angle energies, Van der Waals energiesor/and electrostatic energies.These results indicate that duplexes containing mismatches are considerably destabilized (Figure 5) compared with their correctly paired parent the extent being dependent on the base composition and sequence of the oligonucleotide as well as on the type and location of the mismatch.The mismatch of DNA leads to alterations of amino acid properties andcan cause a change in protein structure [45, 46]. Consequently, SNP may affect enzyme activity through the modification of protein structure and function [47].

Ourresults evaluate the first case of an oligonucleotide split probe system based on excimer fluorescence emission for detection of CYP3A4*1B single nucleotide polymorphism. Further studies will be necessary to understand the details of thesplit probe system structure which determine the formation of the excimer for CYP3A4 single nucleotide polymorphism. Based on fluorescence and spectrophotometric results, the split probe system is selective enough to detect single base mutations of CYP3A4*1B with good sensitivityand thereforecould be used to detect other mutations using an excimer system.
The authors gratefully acknowledge the support and valuable suggestions obtained from Sir Khaled AB Diab (Judicial Expertise and Research Centre, Tripoli, Libya).
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
